Nowadays, the ultrafast sciences paradigm is changing from working on the femtosecond to a shorter sub-femtosecond or attosecond timescale. Such research attracts scientists’ attention as it allows studying different atomic and molecular processes on the scale of the fastest electronic motion. I have already touched on the subfemtosecond spin-flip dynamics simulations in the highly excited states in my blog. In a new article, we continued research in this direction and have asked a question: are the ultrafast spin dynamics in the 2p-core-excited states a predominantly atomic process or the chemical environment plays a crucial role?
Vladislav Kochetov, Huihui Wang, Sergey I. Bokarev Effect of chemical structure on the ultrafast spin dynamics in core-excited states J. Chem. Phys. (2020) 153, 044304.
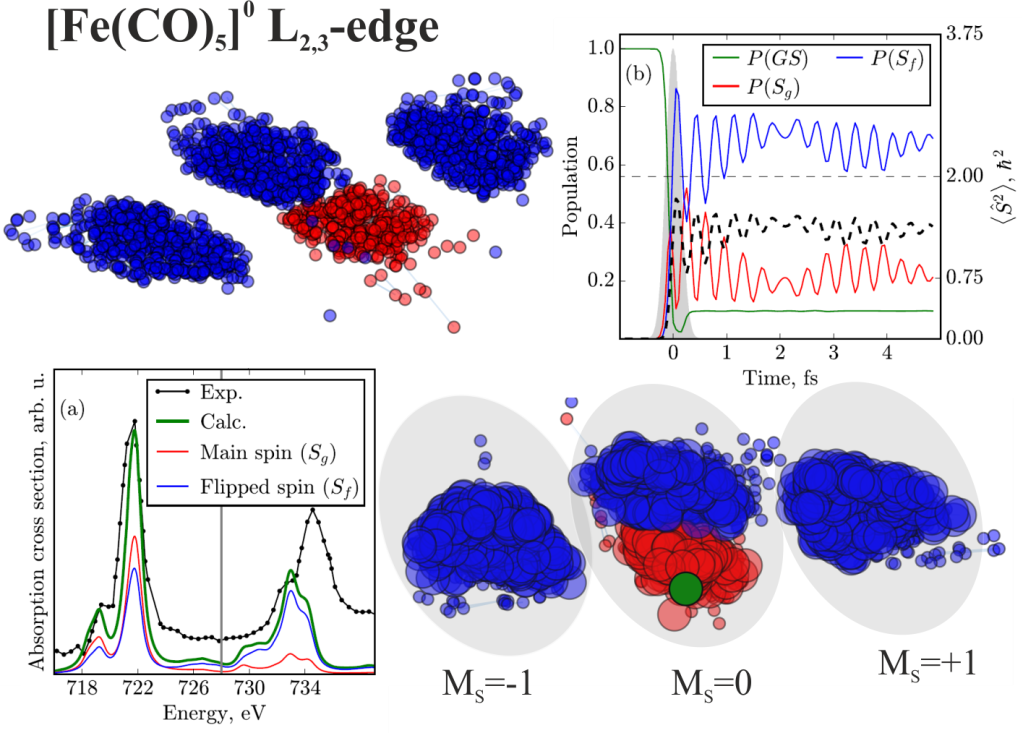
From a general viewpoint, such dynamics are rooted in preparing the superposition of the spin-orbit split states with 2p3/2 and 2p1/2 core holes. Thus, it should be nearly an atomic process, and the strength of the spin-orbit coupling of the hole-bearing atom is decisive. However, the systematic theoretical study of several transition metal complexes of titanium, chromium, iron, and nickel has demonstrated that these simple considerations do not hold.
The ligands attached to the same ion appeared to play only a minor role; of course, if one does not change, e.g., all weak-field ligands to strong-field ones. (In the latter case, the dynamics may change qualitatively.) In turn, the central metal ion’s influence is notably more pronounced but does not correlate with the strength of spin-orbit coupling. For instance, the titanium complex demonstrates an efficient spin-flip, whereas the nickel one does not. It is despite a three times larger coupling constant for nickel.
The dynamics are susceptible to the energetic distribution of states with different multiplicity and their “availability” for the excitation by an ultrashort pulse. The figure above shows that singlet (red) and triplet (blue) states cluster according to spin-orbit interaction and dipole absorption strength. Those nodes which are larger correspond to states which are involved in the dynamics. Those with small nodes are unaffected. Decisive for the efficiency is the ratio between the number of involved states with flipped spin (big blue nodes) and the number of involved states with the ground state spin (big red nodes). The influence of vibrations, which are also inherent to the chemical structure of the complex, was found to be negligible.
This work adds to the understanding of the spin-flip dynamics mechanism and suggests some ways to decrease the computational effort and thus include more states in future simulations.




You must be logged in to post a comment.